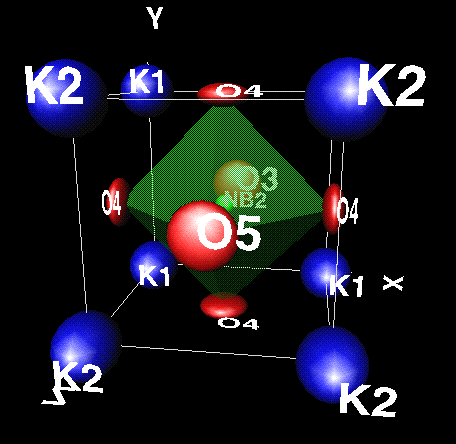
 xtal-3c was made to be used with a minimum of data or instruction. Typing just xtal-3c will
tell you how to use the program, and xtal-3c x will generate a
sample VRML structure file as illustrated on the left (click the image to
obtain it full size). The program
then waits for instructions, which can be as simple as g to draw the structure: a list of instructions
is obtained by typing ? or a carriage return. Note how ellipsoids have been drawn to illustrate thermal vibration amlitudes,
but of course sperical atoms can also be chosen.
xtal-3c was made to be used with a minimum of data or instruction. Typing just xtal-3c will
tell you how to use the program, and xtal-3c x will generate a
sample VRML structure file as illustrated on the left (click the image to
obtain it full size). The program
then waits for instructions, which can be as simple as g to draw the structure: a list of instructions
is obtained by typing ? or a carriage return. Note how ellipsoids have been drawn to illustrate thermal vibration amlitudes,
but of course sperical atoms can also be chosen.
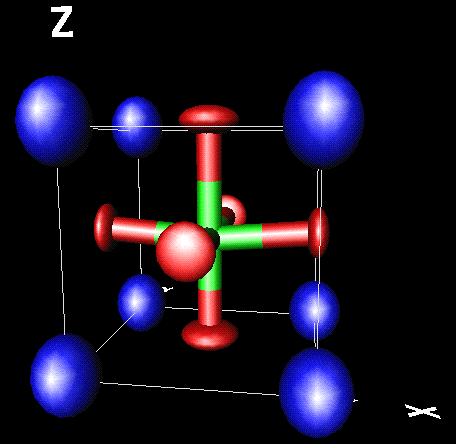 By default xtal-3c will look for bonds between the last two types of atoms in the structure file,
but it can be told instead to use bonds between At1-X and At2-Y with instructions At1-X At2-Y. By
default it will try to construct polyhedrae using a list of atom radii and colours kept in file
xtal-3d.at: this file can be edited by the user, but the user can also change these parameters
on the fly by issuing instructions such as At-X 1,1,0 to draw yellow atoms and polyhedra around At.
Instead of polyhedrae (command p) cylindrical c or simple stick s bonds can be drawn,
or some combination.
By default xtal-3c will look for bonds between the last two types of atoms in the structure file,
but it can be told instead to use bonds between At1-X and At2-Y with instructions At1-X At2-Y. By
default it will try to construct polyhedrae using a list of atom radii and colours kept in file
xtal-3d.at: this file can be edited by the user, but the user can also change these parameters
on the fly by issuing instructions such as At-X 1,1,0 to draw yellow atoms and polyhedra around At.
Instead of polyhedrae (command p) cylindrical c or simple stick s bonds can be drawn,
or some combination.
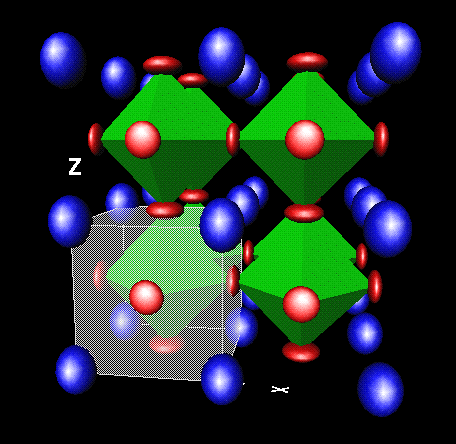 Atoms can be drawn at full size (space filling), as small spheres, or invisible.
Atoms can be labelled, the unit cell drawn as a transparent or semi-transparent box etc., and of course
multiple unit cells can be added in any direction.
Atoms can be drawn at full size (space filling), as small spheres, or invisible.
Atoms can be labelled, the unit cell drawn as a transparent or semi-transparent box etc., and of course
multiple unit cells can be added in any direction.If you are using one of the graphic user interfaces or the WWW version all this becomes obvious, but the basic commands can be entered if prefered, or the commands can be attached directly to the structure file itself.